![]()
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 Hypermobiliteit
Hypermobiliteit
Inleiding
Uw arts heeft u verteld dat u het hypermobiliteitssyndroom hebt. Dit is één van de ruim honderd reumatische aandoeningen. Er komen ongetwijfeld veel vragen op u af. Vragen over de ziekte zelf of over de behandeling.
Op deze website geven we u zoveel mogelijk antwoorden op deze vragen. De informatie is samengesteld op basis van gegevens die we gekregen hebben van artsen, andere behandelaars en mensen met reuma.
Wat is hypermobiliteit?
Hypermobiliteit wil zeggen dat uw gewrichten beweeglijker zijn dan bij andere mensen. Dit komt doordat de gewrichtsbanden erg elastisch en rekbaar zijn. Het is geen ziekte maar een verschijnsel, dat soms wel lastige gevolgen heeft. Men spreekt van het hypermobiliteitssyndroom als u last krijgt van het bewegingsapparaat door te soepele banden. Het gaat dan om spier- en gewrichtsklachten, die niet door een andere bindweefselaandoening veroorzaakt kunnen zijn.
Vooral de gewrichten van de vingers, knieën en wervelkolom kunnen te soepel zijn. Over het algemeen zijn de andere gewrichten dan ook extra soepel.
Hypermobiliteit komt vrij veel voor: naar schatting heeft ongeveer 4 tot 7% van de bevolking er last van. Lang niet alle mensen met hypermobiliteit krijgen klachten. Te soepele gewrichten komen vooral voor bij jongere mensen omdat de elasticiteit van de gewrichtsweefsels afneemt als u ouder wordt. Bij mannen gebeurt dat meestal vanaf het 25e jaar, bij vrouwen vanaf het 45e jaar. Vrouwen hebben overigens sowieso soepelere banden dan mannen. Tijdens de zwangerschap neemt de mobiliteit van de gewrichten nog eens toe. De soepelheid van de gewrichten is niet bij ieder mensenras hetzelfde.
De verschijnselen
Bij het hypermobiliteitssyndroom krijgt u last van het bewegingsapparaat doordat uw banden te soepel zijn. Meestal begint het met vage klachten, zoals spierpijn en pijn in de gewrichten. Daarna ontstaan vaak lage rugklachten. U kunt ook pijn krijgen met traplopen of andere activiteiten die de knieën en enkels belasten. Later ontstaan vaak op meer plaatsen klachten. De gewrichten kunnen opzwellen als er een vochtophoping ontstaat. De knieschijf schiet makkelijker uit de kom dan bij mensen die niet hypermobiel zijn.
Te soepele bekkenbanden kunnen pijnklachten geven die lijken op bekkeninstabiliteit. Dit zijn onder meer lage rugpijn, pijn in het stuitje en pijn bij het opstaan (startpijn). Sommige mensen hebben voortdurend klachten omdat de banden steeds overbelast worden, andere hebben acute, kortdurende klachten.
Jonge kinderen met hypermobiliteit hebben een verhoogde kans op heupluxatie (de kop van het dijbeen zit dan niet goed in de kom van de heup). Het lijkt er ook op dat hypermobiele kinderen zich motorisch net wat langzamer ontwikkelen dan andere kinderen. Zij gaan bijvoorbeeld vaak wat later lopen. De motorische achterstand hebben ze echter voor hun derde jaar ingehaald.
Te soepele gewrichten geven over het algemeen geen ernstige problemen. U kunt er echter wel last van hebben omdat uw gewrichten gevoeliger zijn voor overbelasting. De kans is wat groter dat u uw enkels, ellebogen of schouders verstuikt tijdens het sporten. Ook bij andere activiteiten waarbij u grote bewegingen maakt kan dit gebeuren. Door hypermobiliteit hebt u ook een wat verhoogd risico op een lichte houdingsafwijking in de wervelkolom (scoliose). Daardoor kunt u bijvoorbeeld rug- of nekpijn krijgen.
Bij sommige beroepen en hobby’s zijn soepele gewrichten een voordeel, zoals balletdansen, turnen of muziekmaken. In deze beroepen zijn dan ook relatief veel mensen met hypermobiliteit.
Hoe wordt de diagnose gesteld?
- De volgende tests worden gebruikt om hypermobiliteit vast te stellen:
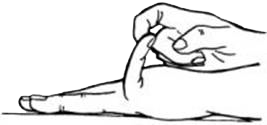
- overstrek de pinken 90° of verder;
- leg de duimen tegen de voorarm;
- overstrek de ellebogen ongeveer 10°
- overstrek de knieën ongeveer 10°
- buig voorover en leg de handen plat op de grond zonder de knieën te buigen.
Kunt u vijf of meer gewrichten op deze manier overstrekken, dan is er sprake van
hypermobiliteit. Laboratoriumonderzoek en bloedonderzoek leveren geen
afwijkende waarden op bij hypermobiliteit. Te soepele banden zijn ook niet op
röntgenfoto’s te zien. Het hypermobiliteitssyndroom, waarbij u allerlei klachten krijgt
door hypermobiliteit, wordt wel verward met bepaalde reumatische aandoeningen.
Zo krijgen sommige patiënten de diagnose fibromyalgie terwijl ze eigenlijk
hypermobiel zijn.
Bij extreme hypermobiliteit zal de arts onderzoeken of er een onderliggende ziekte
is die de hypermobiliteit veroorzaakt. Extreme hypermobiliteit is echter zeldzaam.
Behandeling
Sommige spieren kunnen een deel van het werk van de gewrichtsbanden opvangen. Een fysiotherapeut kan u helpen met oefeningen die bepaalde spiergroepen extra verstevigen. Bij ernstige klachten zijn orthesen soms geschikt. Dit zijn ondersteunende hulpmiddelen, bijvoorbeeld een polscorset, een SI-band of een halskraag.
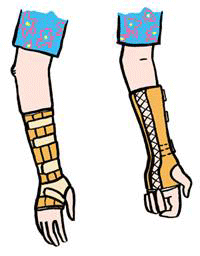 Sommige hypermobiele mensen hebben veel baat bij speciale zooltjes in de schoenen (hiervoor kunt u naar een podo-orthesioloog of orthopedisch instrumentenmaker) of een eenvoudige hakverhoging die knieproblemen tegengaat. U kunt eventueel pijnstillers gebruiken als ondersteuning van de behandeling. Bedenk daarbij wel dat pijnstillers alleen de symptomen bestrijden.
Sommige hypermobiele mensen hebben veel baat bij speciale zooltjes in de schoenen (hiervoor kunt u naar een podo-orthesioloog of orthopedisch instrumentenmaker) of een eenvoudige hakverhoging die knieproblemen tegengaat. U kunt eventueel pijnstillers gebruiken als ondersteuning van de behandeling. Bedenk daarbij wel dat pijnstillers alleen de symptomen bestrijden.
Bandages kunnen de kans op klachten en blessures verminderen. Het is erg onverstandig een bandage te gebruiken om een sport te kunnen (blijven) doen, terwijl u deze sport zonder een bandage niet zonder klachten kunt volhouden.
Wat kunt u zelf doen?
Om te voorkomen dat u klachten krijgt kunt u het beste enkele preventieve maatregelen nemen. Voorkom overbelasting van uw gewrichten. U kunt beter geen sporten met een wedstrijdelement doen. Ook sporten met hoge piekbelastingen zijn minder geschikt. Dit zijn sporten waarbij u veel snelle bewegingen maakt en steeds kort van houding verandert, zoals tennis, boksen, squash, basketbal en skiën.
Werk
Kleine aanpassingen op de werkplek kunnen een aanzienlijke vermindering van klachten geven. Herhaalde bewegingen (monotoon werk) en werken boven het hoofd dienen zoveel mogelijk te worden vermeden.
Kijk ook op de website:
www.hypermobiliteitssyndroom.nl
Wat is EDS,
het Ehlers-Danlos
syndroom?
Inleiding
Het syndroom van Ehlers-Danlos bestaat uit een groep van zes erfelijke bindweefsel aandoeningen. EDS is een zeldzame, erfelijk bepaalde aandoening. Met name de huid, gewrichtsbanden, kapsels en organenwanden kunnen aangedaan zijn.
Het woord 'syndroom' betekent dat een combinatie van meerdere ziekteverschijnselen gezamenlijk één ziektebeeld vormen.
De belangrijkste kenmerken zijn een zeer rekbare huid, te lenige gewrichten en het vaker optreden van blauwe plekken.
Het syndroom komt bij, naar schatting, 1 op de 5.000 mensen voor, wat neerkomt op ongeveer 3.000 mensen in Nederland.
Wat is EDS?
Het Ehlers-Danlos syndroom (EDS) is een erfelijke aandoening waarbij het bindweefsel niet goed is aangelegd. Bind- of steunweefsel zit overal in ons lichaam waar steun gegeven moet worden. Dit betekent dat het een belangrijk bestanddeel is van bijvoorbeeld botten en van de banden die gewrichten op hun plaats houden. Maar het zorgt er ook voor dat de huid vast zit aan de onderliggende laag en dat de tanden in hun kassen blijven zitten. De samenstelling van het bindweefsel hangt af van de functie die het in ons lichaam vervult. Een stoornis in het bindweefsel van de gewrichtsbanden die moeten zorgen dat gewrichten stevig in elkaar zitten, geeft andere klachten dan een stoornis in het bindweefsel dat de huid stevigheid geeft. Dit is dan ook de reden dat er verschillende types van het Ehlers-Danlos syndroom worden onderscheiden. Hoeveel mensen aan dit syndroom lijden is niet bekend.
Wat is de oorzaak van de ziekte?
Alle types Ehlers-Danlos zijn erfelijk. Ze kunnen echter op een verschillende manier overerven: Een dominant erfelijke manier, waarbij de aandoening direct van ouder op kind overerft. Ook kan de overerving van het syndroom plaatsvinden op een recessieve manier, waarbij de ouders ongemerkt drager zijn van een verborgen overervende ziekte-aanleg en waarbij een kind de ziekte krijgt als het van beide gezonde ouders deze ziekte-aanleg erft. Er is een geslachtsgebonden overerving waarbij de aandoening alleen bij jongens voorkomt. En je kunt het als eerste in de familie krijgen. Dan is er sprake van een spontane mutatie.
Hypermobiliteitstest van Bulbena
Deze test wordt weergegeven met diverse foto's met een medisch karakter. Als je hiervoor gevoelig bent, kan het beter zijn dit gedeelte over te slaan.
Wilt u deze foto's zien, klik dan hier.
Huidafwijkingen, variërend in ernstige of minder ernstige mate, komen in 100% van de gevallen voor bij Ehlers-Danlos. Het is belangrijk niet te proberen of je huid overrekbaar is, laat dit altijd aan een arts over.
Voor meer informatie kunt u contact opnemen met:
de Vereniging van Ehlers Danlos Patienten:
Stationsplein 6
3818 LE Amersfoort
0900 - VED INFO
(0900 - 463 6833)
info@ehlers-danlos.nl
het syndroom van Marfan
Wat is Marfan?
 Het syndroom van Marfan is een aangeboren en erfelijke afwijking van het bindweefsel. Dit weefsel komt op veel plaatsen in het lichaam voor. De belangrijkste Marfanverschijnselen zijn te zien aan hart, bloedvaten, ogen en skelet. Syndroom geeft aan dat het gaat om een verzameling van afwijkingen die samen en juist in deze combinatie voorkomen. Alle verschijnselen samen zijn te verklaren vanuit één oorzaak. Marfan is de naam van een Franse kinderarts die aan het eind van de vorige eeuw als eerste een patiëntje beschreef met die aandoening. Daarom is de ziekte naar hem vernoemd. Het syndroom van Marfan komt overal ter wereld voor. 1 op de 10.000 mensen heeft deze aandoening. Het komt even vaak voor bij mannen als bij vrouwen. In Nederland zijn ongeveer 1500 mensen waarvan is aangetoond dat zei deze aandoening hebben.
Het syndroom van Marfan is een aangeboren en erfelijke afwijking van het bindweefsel. Dit weefsel komt op veel plaatsen in het lichaam voor. De belangrijkste Marfanverschijnselen zijn te zien aan hart, bloedvaten, ogen en skelet. Syndroom geeft aan dat het gaat om een verzameling van afwijkingen die samen en juist in deze combinatie voorkomen. Alle verschijnselen samen zijn te verklaren vanuit één oorzaak. Marfan is de naam van een Franse kinderarts die aan het eind van de vorige eeuw als eerste een patiëntje beschreef met die aandoening. Daarom is de ziekte naar hem vernoemd. Het syndroom van Marfan komt overal ter wereld voor. 1 op de 10.000 mensen heeft deze aandoening. Het komt even vaak voor bij mannen als bij vrouwen. In Nederland zijn ongeveer 1500 mensen waarvan is aangetoond dat zei deze aandoening hebben.
Uit het medisch handboek:
Symptomen
 Het gen voor het syndroom Marfan is dominant, maar niet iedereen die de aandoening erft, wordt in gelijke mate aangetast. Mensen met dit syndroom zijn langer dan gemiddeld en dan in hun familie gebruikelijk is. Hun armweidte (de afstand tussen vingertoppen van linker-en rechterhand bij opzij gestrekte armen) is groter dan hun lengte. Ze hebben lange, dunne vingers. Hun borstbeen (sternum) is vaak misvormd en wijst naar buiten of juist naar binnen. De gewrichten kunnen zeer beweegbaar zijn. Platvoeten en een bochel met een abnormale kromming van de wervelkolom (kyfoscoliose) komen ook veel voor, evenals breuken. De persoon heeft meestal weinig onderhuids vet en vaak een verhoogd gehemelte. De lens van beide ogen kan zijn verschoven. Met behulp van een oftalmoscoop kan een arts dan in de pupil de rand van de verschoven lens zien.
Het gen voor het syndroom Marfan is dominant, maar niet iedereen die de aandoening erft, wordt in gelijke mate aangetast. Mensen met dit syndroom zijn langer dan gemiddeld en dan in hun familie gebruikelijk is. Hun armweidte (de afstand tussen vingertoppen van linker-en rechterhand bij opzij gestrekte armen) is groter dan hun lengte. Ze hebben lange, dunne vingers. Hun borstbeen (sternum) is vaak misvormd en wijst naar buiten of juist naar binnen. De gewrichten kunnen zeer beweegbaar zijn. Platvoeten en een bochel met een abnormale kromming van de wervelkolom (kyfoscoliose) komen ook veel voor, evenals breuken. De persoon heeft meestal weinig onderhuids vet en vaak een verhoogd gehemelte. De lens van beide ogen kan zijn verschoven. Met behulp van een oftalmoscoop kan een arts dan in de pupil de rand van de verschoven lens zien.
 De patiënt kan zeer bijziend zijn en het netvlies (het lichtgevoelige gebied achter in het oog) kan loslaten. Een zwakke plek in de aortawand kan een geleidelijke verwijding van de aorta veroorzaken, waardoor een aneurysma (uitstulping van de vaatwand) ontstaat. Tussen de lagen van de vaatwand kan bloed komen (aortadissectie) of het aneurysma kan scheuren, waardoor een zeer ernstige bloeding ontstaat. Naarmate de aorta wijder wordt, kan de aortaklep, die zich tussen het hart en de aorta bevindt, beginnen te lekken (aortaregurgitatie).De mitralisklep, die zich tussen de linkerboezem en linkerkamer bevindt, kan gaan lekken of uitstulpen in de linkerboezem. In de longen kunnen vochthoudende blaasjes (cysten) ontstaan. Als deze openbarsten, ontstaat er een pneumothorax (lucht in de ruimte rond de longen), waardoor ademhalen moeilijk wordt.
De patiënt kan zeer bijziend zijn en het netvlies (het lichtgevoelige gebied achter in het oog) kan loslaten. Een zwakke plek in de aortawand kan een geleidelijke verwijding van de aorta veroorzaken, waardoor een aneurysma (uitstulping van de vaatwand) ontstaat. Tussen de lagen van de vaatwand kan bloed komen (aortadissectie) of het aneurysma kan scheuren, waardoor een zeer ernstige bloeding ontstaat. Naarmate de aorta wijder wordt, kan de aortaklep, die zich tussen het hart en de aorta bevindt, beginnen te lekken (aortaregurgitatie).De mitralisklep, die zich tussen de linkerboezem en linkerkamer bevindt, kan gaan lekken of uitstulpen in de linkerboezem. In de longen kunnen vochthoudende blaasjes (cysten) ontstaan. Als deze openbarsten, ontstaat er een pneumothorax (lucht in de ruimte rond de longen), waardoor ademhalen moeilijk wordt.
De kans op complicaties is sterk afhankelijk van de ernst van de afwijkingen. Het grootste gevaar is een plotselinge scheur in de aorta, wat al snel dodelijk kan zijn. De kans op een gescheurde aorta is groter bij het uitoefenen van actieve sporten.
Diagnose en behandeling
Als een buitengewoon lange en slanke persoon met een van de genoemde
kenmerkende symptomen bij de dokter komt, moet deze bedacht zijn op het
syndroom van Marfan. Veel mensen die aan dit syndroom lijden, vertonen echter
nooit symptomen waardoor de aandoening niet bij hen wordt herkend.
Het belangrijkste doel van de behandeling is het voorkomen van problemen met de
bloedvaten en ogen. De ogen worden een keer per jaar onderzocht. Zodra zich
gezichtproblemen voordoen, moet een patiënt met het syndroom van Marfan
onmiddellijk een arts raadplegen.
Om verwijding (dilatatie) en dissectie (bloed tussen de lagen van de vaatwand) van
de aorta te voorkomen, kan propanol worden gebruikt, een middel dat de kracht
van de bloedstroom vermindert. Een verwijding in de aorta kan soms operatief
worden verholpen.
Kinderen met het syndroom van Marfan kunnen zeer lang worden. Een arts kan zeer
lange meisjes een hormoonbehandeling voorschrijven (oestrogeen en
progresteron). Hiermee wordt meestal begonnen op tienjarige leeftijd om de
pubertijd in te leiden en daardoor de groei te stoppen.
Wat veroorzaakt het Marfan syndroom?
Een enkel abnormaal gen veroorzaakt het Marfan syndroom. Meestal is dit gen doorgegeven door een ouder die de drager is. Soms, ongeveer een kwart van alle Marfan patiënten, wordt het syndroom veroorzaakt als gevolg van spontane mutatie.
Het Marfan syndroom is autosomaal dominant. Met als gevolg dat als iemand met het syndroom een kans van 50-50 heeft dat een nakomeling ook Marfan heeft.Pas sinds 1991 begint meer bekend te worden over de oorzaak van Marfan. We weten nu dat het een stoornis is in het bindweefsel. Meer precies: één van de eiwitten die nodig zijn voor de aanmaak van bindweefsel, is niet helemaal goed samengesteld. De erfelijke code voor het aanmaken van het eiwit fibrilline bevat een "bouwfout". Het fibrilline ziet er hierdoor ietwat anders uit dan eigenlijk zou moeten.Bindweefsel dat met dit eiwit gemaakt wordt, is door de fout minder goed van samenstelling. Het werkt ook niet helemaal zoals dat zou moeten. Omdat bindweefsel overal in het lichaam voorkomt, kan zo'n verandering op veel plaatsen problemen opleveren.
Iemand met Marfan zal minstens enkele van de volgende kenmerken hebben:
hart en bloedvaten
 - geleidelijk toenemende verwijding van de grote lichaams-slagader (aorta)
- geleidelijk toenemende verwijding van de grote lichaams-slagader (aorta)
- stoornis aan de hartkleppen
ogen
- verplaatsing van de ooglens
- bijziendheid
- verhoogde druk in het oog
- netvlies loslating
skelet
- afwijking in de vorm van de borstkas
- naar verhouding lange armen en benen
- opvallende lichaamslengte
- slappe gewrichtsbanden, met als gevolg overbeweeglijke gewrichten, platvoeten, scoliose
overige
- weinig onderhuids vet
- 'klap-long' en liesbreuk
- onderhuidse striemen (striae)
- verwijding van de ruimte waarin zich de vloeistof van het ruggemerg bevindt
Diagnose en behandeling
Niet elke patiënt heeft precies dezelfde kenmerken, zelfs niet binnen één familie. Soms treden verschijnselen pas in de loop van de tijd op. De diagnose kan daardoor lastig te stellen zijn. Er bestaat nog geen laboratoriumtest voor. Een team van artsen (cardioloog, oogarts, orthopeed, erfelijkheidsdeskundige) zal de patiënt moeten onderzoeken. Deze onderzoeken zijn niet pijnlijk: echo-opnames van het hart, onderzoek van het oog met een speciale lamp, röntgenfoto's, etc.
Steeds wordt gezocht naar de typische kenmerken en verschijnselen die bij Marfan horen. Over het stellen van de diagnose zijn door de artsen internationale afspraken gemaakt. Op enkele plaatsen in Nederland bestaan inmiddels speciale Marfanteams en Marfan-poliklinieken.
Het is belangrijk dat het syndroom van Marfan zo vroeg mogelijk wordt vastgesteld! Grote problemen kunnen dan vaak worden uitgesteld of voorkomen. Enkele leefregels, maar vooral regelmatige medische controles, zijn letterlijk van levensbelang. Met name de geleidelijke wijder wordende lichaamsslagader (aorta) moet worden gecontroleerd. Dit bloedvat zou namelijk kunnen scheuren als het te wijd wordt. Tijdig operatief ingrijpen voorkomt deze - vaak dodelijke - complicatie.
Als de diagnose syndroom van Marfan gesteld is, is het belangrijk om ook de ouders, broers/zussen en kinderen van de patiënt te onderzoeken. Misschien hebben zij de aandoening ook, en dan moeten ook zij zorgvuldig worden begeleid en gecontroleerd
Onderstaande poliklinieken zijn gericht op Marfan
| Marfan Polikliniek Amsterdam |
Coördinator: Dr. R. Hennekam (klinisch geneticus) Academisch Medisch Centrum (AMC) Meibergdreef 15 1105 AZ Amsterdam Onderzoeksdagen: volwassenen: elke vierde woensdag van de maand; afspraken: secretariaat afdeling klinische genetica; tel: 020 5665281 (dagelijks van 8.30 –12.30) kinderen: elke derde (en vijfde) woensdag van de maand; afspraken: afdeling Kindergeneeskunde tel: 020 5663386 |
| Marfan Polikliniek Nijmegen |
Coördinator: Dr. F. Boers (internist) Academisch Ziekenhuis Nijmegen St. Radboud Afdeling Alg. Interne Geneeskunde (541) Postbus 9101 6500 HB Nijmegen Onderzoeksdagen volwassenen: iedere maandag afspraken: via secretaresse mevr. M.Vink-Knuist; tel: Dinsdag: 024 3618950 tel: Donderdag en vrijdag: 024-3614534 Kinderen: elke eerste maandag van de maand; afspraken: via secretariaat polikliniek kindergeneeskunde; tel: 024 3614427 |
| Marfan polikliniek Groningen |
Coördinator: Dhr E.H. Sikkens (maatschappelijk werker) Academisch Ziekenhuis Groningen (AZG) A. Deursinglaan 4 9713 AW Groningen onderzoeksdagen: kinderen en volwassenen iedere vierde dinsdag van de maand (behalve juli, augustus en december) afspraken: via dhr. Sikkens tel: 050-3632929 |
| Marfan poli-kliniek LUMC - Leiden |
iedere Dinsdag middag op cardiologie, bij Dr.Haverkamp. afspraken en info. bel 071-5263728 (dr. Krauss is gepensioneerd sinds juni 2005) |
Vragen en of opmerkingen kunnen gestuurd worden naar:
meike@marfan.nl
Kijk ook op de website:
www.marfan.nl
Bronnen:
- Reumapatiëntenbond
- de Vereniging van Ehlers Danlos Patiënten
- Contactgroep Marfan Nederland
Met dank aan de Vereniging van Ehlers Danlos Patiënten en Contactgroep Marfan Nederland voor het beschikbaar stellen van hun informatie.
Voor de inhoud van dit artikel verwijzen wij naar de bronnen en draagt de RPV ’s-Hertogenbosch e.o. dan ook geen verantwoording!